{kind=link}
{kind=link}
{kind=link}
{kind=link}
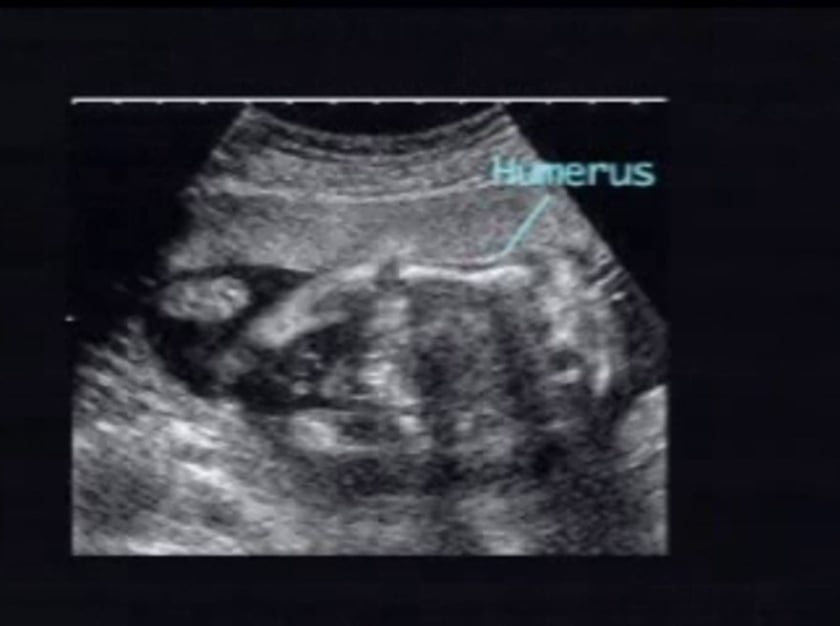
osteogenesis imperfecta iii
this fetus displays typical appearances of mild bone shortening with long bone fractures and moderately decreased mineralization. the spine and ribs are relatively normal. the iliac bones appear undermineralized. tibial bowing is prominent.